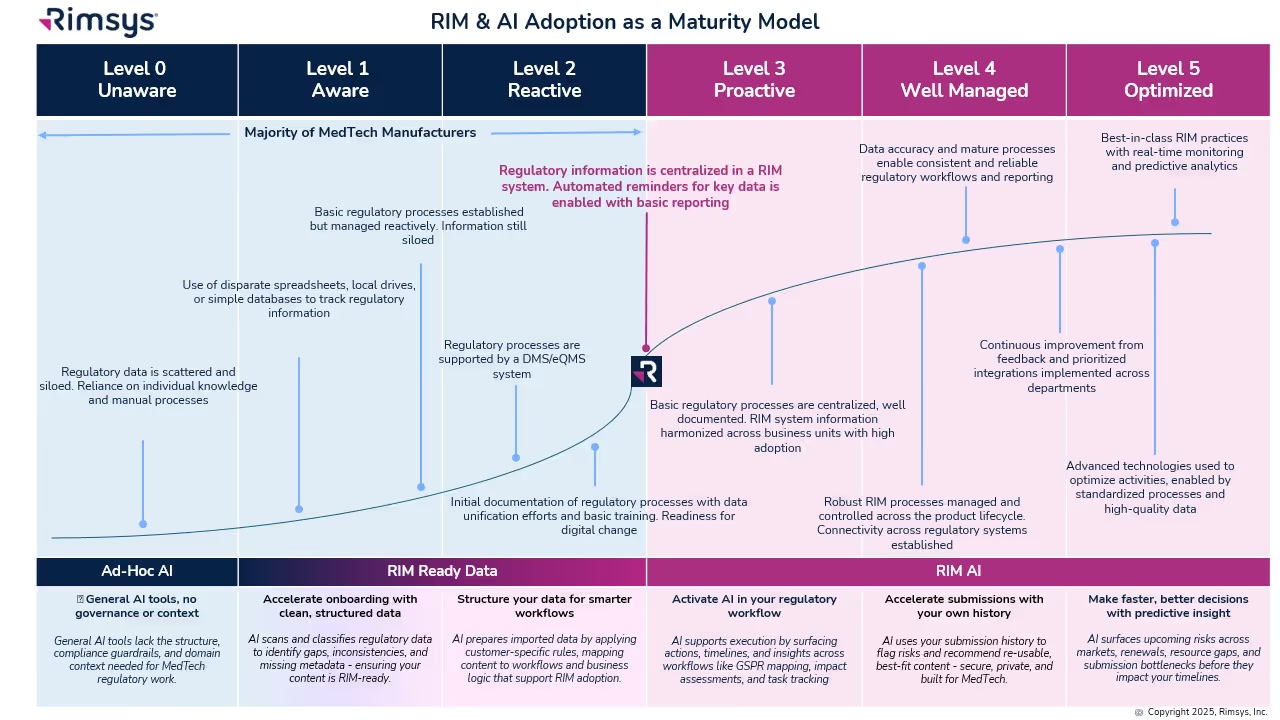
History
In September of 2018, the FDA took their first step in their commitment to improve the process of a 510(k) electronic submission. This commitment was established as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) that was enacted on July 9, 2012, amending the FD&C Act by adding section 745A.
Based on the FDA’s experience with the original eSubmitter process, the Center for Biologics Evaluation and Research (CBER) group piloted a slightly different program and called it the Electronic Submission Template and Resource Pilot Program, thus creating eSTAR. This second attempt at electronic submissions consists of a collection of questions, text, and prompts within a PDF template that guides a user to a ‘complete’ 510(k) electronic submission.
The FDA’s goal of this process is to enhance the quality of submissions by helping to ensure consistent, quality, comprehensive data for the Center for Disease and Radiological Health (CDRH) premarket review. With a standardized format in place, submitters of a 510(k) or De Novo can ensure that their submissions are complete and premarket reviews will be more efficient.
eSTAR timeline
.avif)
eSTAR Requirements
Currently, eSTAR is a voluntary process that will become mandatory starting October 1, 2023. It is free to use by medical device applicants wishing to submit a 510(k) or a De Novo to the CDRH, but is not to be used for combination products. Although it’s currently free to use, the standard submission fees for 510(k) and De Novo still apply.
A Refuse to Accept (RTA) review (a preliminary review used to ensure the submission is complete) will not be conducted on submitted eSTAR templates as the eSTAR template replaces this checklist and will state if the template is complete or incomplete on the first page. If the eSTAR does not have a completed status, it will not be reviewed by the FDA.
Guidance Information
The FDA has provided final guidance documentation in “Providing Regulatory Submissions for Medical Devices in Electronic Format — Submissions Under Section 745A(b) of the Federal Food, Drug, and Cosmetic Act” (referred to as the “745A(b) device parent guidance”). This guidance document provides a process for the development of templates to facilitate the preparation, submission, and review of regulatory submissions for medical devices solely in electronic format.
The FDA then provided final guidance in “Electronic Submission Template for Medical Device 510(k) Submissions.” There are exemptions listed in the guidance document that may allow users in certain situations to be exempted from eSTAR.
eSTAR Templates
The two templates provided by the FDA are PDF forms that can be filled out and saved, with attachments. The image below shows the device description page of the non-IVDR form. When using the form, red bars will appear in front of any required questions that have not yet been answered. Bars will turn green when all associated questions in a section have been completed. Gray bars indicate an optional question.

As questions are answered, additional dropdowns may appear that can require users to add additional information. For example, in the Pre-Submission Correspondence and Previous Regulatory Interaction Section within the non-IVD template, if you Select “Yes” the PDF will then add a section where you will need to add the Submission, Submission Number, and copies of the correspondence.

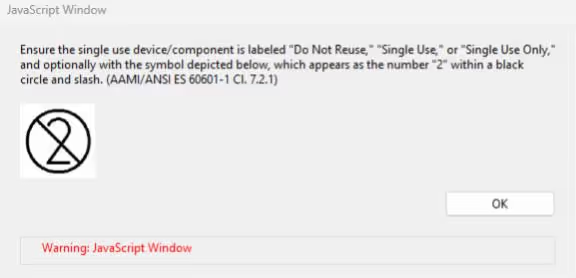
Pop-ups with useful information may also appear. For example, within the non-IVD template, in the General Device Characteristics Section, if a user selects that a device is a single use device, non-sterile or packaged as sterile, a Java Script window will appear with FDA guidance.
A note on embedded attachments: Any embedded attachments may only be used once per eSTAR PDF. You may need to “break up” documentation to fit the eSTAR template requirements if your testing reports cover multiple tests in one document.
What’s involved in a submission?
There are currently two processes for submission:
- The Customer Collaboration Portal
- Mailing the eCopy (CD, DVD or USB drive) containing the eSTAR PDF
First, effective July, 2022, The Customer Collaboration Portal was made available to existing users of the portal if they are the official correspondent of an organization and have also received the FDA email letting them know that they’re eligible.
The second method consists of a printed cover letter with an accompanying eCopy (CD, DVD or USB drive) containing the eSTAR PDF. Note that if the PDF submission exceeds 1 GB in size, it could delay the process, so high-resolution videos and images should only be included when necessary.
- Download the specific eSTAR PDF template for an In Vitro or Non-In Vitro device (see "Current eSTAR versions" section on this linked page).
- Fill out the template accordingly but note that it is only used for constructing - not submitting - your submission.
- The eSTAR does not need to comply with the FDA’s eCopy Guidance document, however, any additional files that are provided with the eSTAR PDF submission will need to comply with it. The FDA strongly recommends not providing additional files with the eSTAR PDF, though, as you already will be embedding your documents within the eSTAR template itself.
- You do not need to provide an Indications for Use page, the Premarket Review Submission Cover Sheet, or a Declaration of Conformity (if applicable) with your submission since they are already built into the eSTAR PDF. An additional option is to also build the 510(k) summary within the template itself instead of writing one.
An eSTAR submission will contain a larger number of files (as they will be embedded in the PDF eSTAR template) than traditional submissions, which are usually combined into one PDF file. We recommend preparing your entire submission before filling out the eSTAR template to minimize the need for reworking the submission.
A note on 510(k) product change submissions: The eSTAR template requires that all subsections be completed, so if you are submitting a 510(k) to address a product change, you will want to include a justification for not including original information in required sections that did not change. Otherwise, all submitted information will be reviewed again, even if it does not differ from the original submission.
Review Timeline
The review timeline will be similar to the review timeline for a non-electronic submission for a 510(k). The guidance document “The 501(k) Program: Evaluating Substantial Equivalence in Premarket Notification [510(k)]” can offer more details. The same would apply for the De Novo in that you would need to reference the guidance document “De Novo Classification Process (Evaluation of Automatic Class III Designation)” for review timelines.
Tracking the Submission
The FDA secures the information about each submission’s progress so only its Official Correspondent (your company’s submission representative) can view it. You will be able to track the progress of your submission by using the Customer Collaboration Portal (CCP).
- If this is your Official Correspondent’s first time tracking a submission online, the FDA will automatically email a link to create a login password soon after the FDA starts its review.
- The FDA currently displays progress online for Traditional, Abbreviated, and Special 510(k) submissions.
- The FDA will formally notify you of your submission’s status by emailing your Official Correspondent with official actions and requests.
The future of eSTAR
These efforts are part of the FDA’s ongoing commitment to work with the industry and to improve the efficiency of the medical device review process. The final guidance for the 510(k) eSTAR was published this month and will become effective October 1, 2023, for 510(k) and De Novo submissions. In the long term if MUDUFA V is passed with the current requests from the FDA, the process will become a required standard for additional submission types such a PMA and will replace the need to mail eCopies containing eSTAR files through the FDA Collaboration Portal.
Similar posts