
MDSAP - the ultimate guide to the medical device single audit program


This article is an excerpt from The ultimate guide to the medical device single audit program (MDSAP) ebook.
Table of contents
- What is MDSAP?
- History of MDSAP
- Who is responsible for the MDSAP?
- How does an MDSAP audit work?
- Audit sequence
- You got a nonconformity – now what?
- What does an MDSAP audit cost?
- Why choose the MDSAP certification process?
- Potential disadvantages of the MDSAP
- Ready to participate? – Here’s how to get started
- Completing a successful MDSAP audit
The Medical Device Single Audit Program (MDSAP) was designed and developed to allow a single audit of a medical device manufacturer to be applied to all country markets whose regulatory authorities are members of the program. The MDSAP provides efficient and thorough coverage of the standard requirements for medical device manufacturer quality management systems, and requirements for regulatory purposes (ISO 13485:2016). In addition, there are specific requirements of each medical device regulatory authority participating in the MDSAP that must be met:
- Conformity Assessment Procedures of the Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3)
- Brazilian Good Manufacturing Practices (RDC ANVISA 16)
- Medical Device Regulations of Health Canada (ISO 13485:2003)
- Japan Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No 169)
- Quality System Regulation (21 CFR Part 820), and specific requirements of medical device regulatory authorities participating in the MDSAP program.
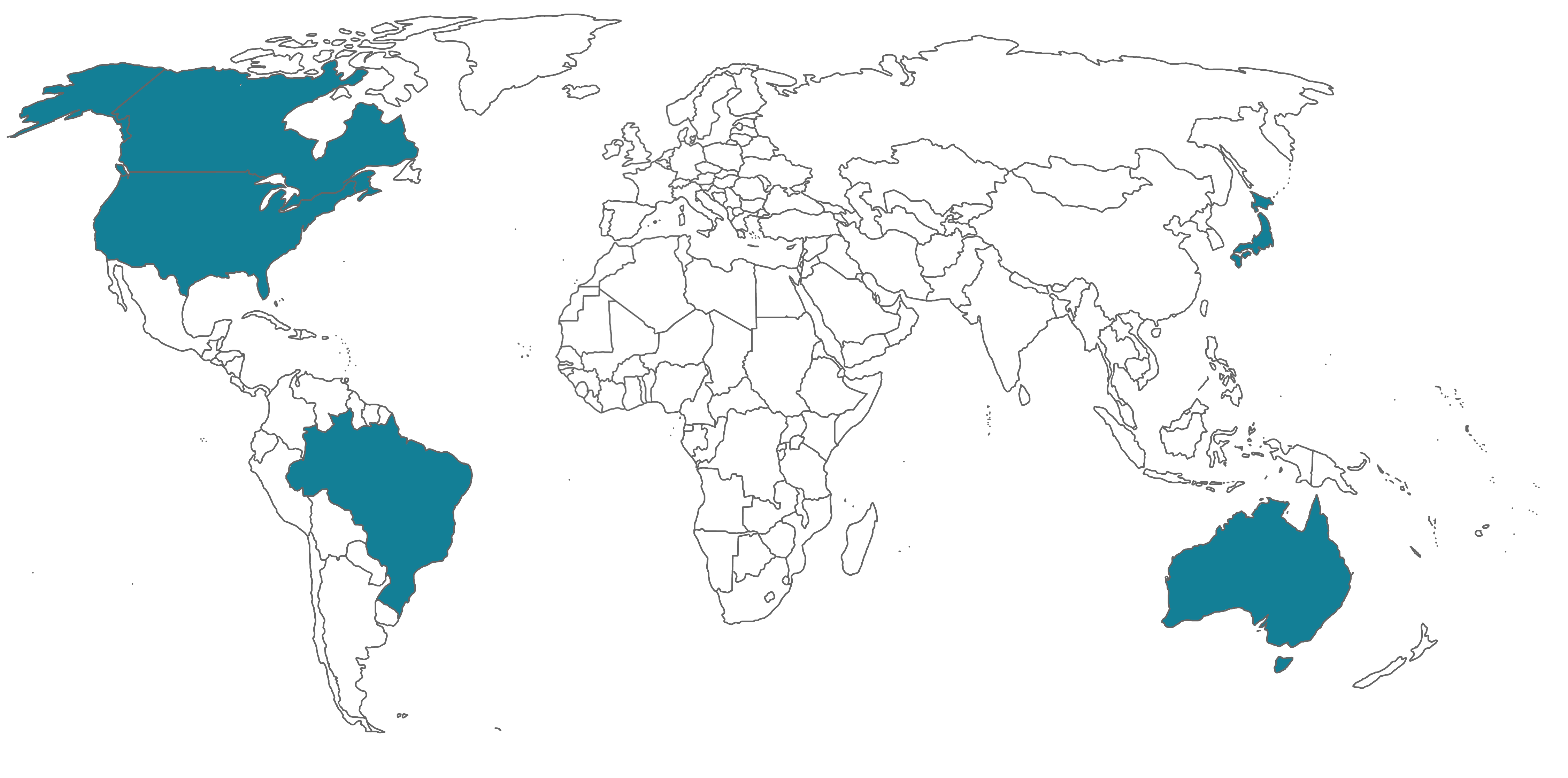
This means that a report from a single MDSAP audit of a medical device manufacturer would be accepted as a substitute for routine inspections by all the member Regulatory Authorities (RAs) across the world. There are currently five participating Regulatory Authorities (RA) representing the following countries: Australia, Brazil, Canada, Japan and the USA.
In April, 2021, the RAs released an “Audit Approach” document (MDSAP AU P0002.006) that combines the formerly separate MDSAP Audit Model and Process Companion documents into a single guidance document. It includes guidance for assessing the conformity of each process and includes an audit sequence, instructions for auditing each specific process, and identifies links that highlight the interactions between the processes.
In March 2012 the US FDA announced that they had approved a final pilot guidance document “Guidance for Industry, Third Parties and Food and Drug Administration Staff: Medical Device ISO 13485:2003 Voluntary Audit Report Submission Pilot Program.” This allowed the owner or operator of a medical device manufacturing facility to be removed from FDA’s routine inspection work plan for 1 year upon completing a ISO 13485:2003 audit. This guidance document went into effect in June 2012, and was intended as an interim measure while a single audit program was being developed.
This pilot program was not very successful and few companies signed up because they did not see any advantage in participating. The manufacturer had to pay for a third party to inspect their facilities, generate a report, and share the inspection results back to the FDA. Many companies were reluctant to contract “someone else” to perform their inspection when they could easily wait for the FDA to conduct an inspection for free.
During its inaugural meeting in Singapore in 2012, the International Medical Device Regulators Forum (IMDRF) appointed a working group to develop a set of documents for a harmonized third-party auditor system. Hence, the “Medical Device Single Audit Program” (MDSAP) was formed. The concept was similar to the FDA’s original idea of creating a third-party auditor to help reduce their workload of performing regulatory audits of medical device manufacturers’ quality management systems. This new approach would consist of a single audit that would review regulatory QMS compliance, conducted by a third-party, who would later be called an Auditing Organization (AO).
From January 2014 to December 2016, five countries participated in a Medical Device Single Audit Program Pilot. In June 2017, a report was generated summarizing the outcomes of prospective “proof- of-concept” criteria established to confirm the success of the program. The outcomes are documented in the final MDSAP Pilot Report and recommended that the program become fully active and open to any manufacturer who requested this type of audit.
The governing body of the MDSAP is the Regulatory Authority Council (RAC), which is composed of two senior managers (and a few other staff members) from each participating RA. They are responsible for executive planning, strategic priorities, setting policy, and making decisions on behalf of the MDSAP International Consortium. The RAC also reviews and approves documents, procedures, work instructions, and more. The mission of the MDSAP International Consortium is to jointly leverage regulatory resources to manage an efficient, effective, and sustainable single audit program focused on the oversight of medical device manufacturers on a global scale.
Other international partners that are involved in the MDSAP include:
MDSAP Observers:
- European Union (EU)
- United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA)
- The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Program
MDSAP Affiliate Members:
- Argentina’s National Administration of Drugs, Foods and Medical Devices (ANMAT)
- Republic of Korea’s Ministry of Food and Drug Safety
- Singapore’s Health Sciences Authority (HSA)
The observers and affiliate members are not the same as the participating member RA’s. The observers simply observe and/or contribute to RAC activities. Affiliate members, on the other hand, are interested in engaging in the MDSAP program and are subject to certain rules. They are only given access to a certain level of information about the manufacturers, audit dates, and information in audit reports.
They are also invited to attend sessions that are open to members, observers, and affiliates only.
Audits can also be conducted by MDSAP participating RAs at any time and for various reasons including:
- "For Cause" due to information obtained by the regulatory authority
- as a follow up to findings from a previous audit
- to confirm the effective implementation of the MDSAP requirements
The purpose of audits conducted by the RAs is to ensure appropriate oversight of the AOs MDSAP auditing activities. The AOs are appointed by the RAs and a list of the currently approved AO’s is published on the FDA website. Most AOs offer a broad range of management system certification services, beyond just medical devices. Manufacturers should verify that prospective AOs are clearly trained and perform MDSAP audits of medical devices.
AOs have the final word as to whether a manufacturer has met the requirements for the MDSAP during the execution of the audit and generation of the associated reports summarizing the results. MSDAP RAC participating RAs have the final decision regarding all development, implementation, maintenance, and expansion activities associated with the program.
Although an unannounced visit by an AO is rare, it can happen in circumstances where high-grade nonconformities have been detected.
To continue reading this eBook including a detailed look at the MDSAP audit process and grading, pros and cons of the approach, and how to get started please register to download the full version.
Similar posts




